Bioinformatics analysis plays a crucial role in discovering patterns existing across DNA sequences using biological data. Relevance Lab’s RLCatalyst Research Gateway offers Nextflow-based pipeline templates that can run all RNA-Seq Analysis steps in one command.
Bioinformatics is a field of computational science that involves the analysis of sequences of biological molecules (DNA, RNA, or protein). It’s aimed at comparing genes and other sequences within an organism or between organisms, looking at evolutionary relationships between organisms, and using the patterns that exist across DNA and protein sequences to elucidate their function. Being an interdisciplinary branch of the life sciences, bioinformatics integrates computer science and mathematical methods to reveal the biological significance behind the continuously increasing biological data. It does this by developing methodology and analysis tools to explore the large volumes of biological data, helping to query, extract, store, organize, systematize, annotate, visualize, mine, and interpret complex data.
The advances in Cloud computing and availability of open source genomic pipeline tools have provided researchers powerful tools to speed up processing of next-generation sequencing. In this blog, we explain leveraging the RLCatalyst Research Gateway portal to help researchers focus on science and not servers while dealing with NGS and popular pipelines like RNA-Seq.
Steps and Challenges of RNA-Seq Analysis
Any Bioinformatics analysis involving next-generation Sequencing, RNA-Seq (named as an abbreviation of “RNA Sequencing”) constitutes of these following steps:
Mapping of millions of short sequencing reads to a reference genome, including the identification of splicing events
Quantifying expression levels of genes, transcripts, and exons
Differential analysis of gene expression among different biological conditions
Biological interpretation of differentially expressed genes
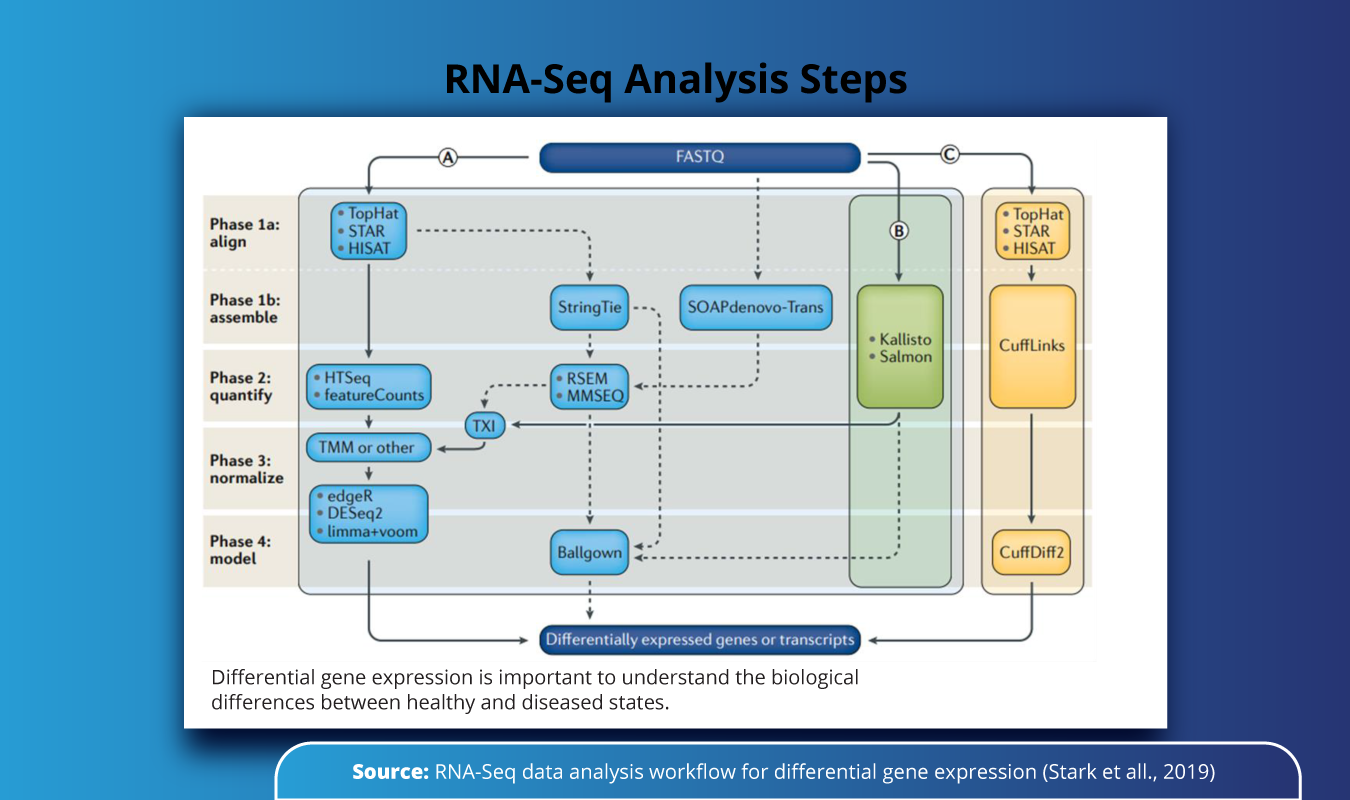
As seen from the figure below, the RNA-Seq analysis for identification of differentially expressed genes can be carried out in one of the three (A, B, C) protocols, involving different sets of bioinformatics tools. In study A, one might opt for TopHat, STAR, and HISAT for alignment of sequences and HTSeq for quantification, whereas the same set of steps can be performed by using Kalisto and Salmon tools (Study B) or in combination with CuffLinks (Study C) all of these yields the same results which are further used in the identification of differentially expressed genes or transcripts.
Each of these individual steps is executed using a specific bioinformatics tool or set of tools such as STAR, RSEM, HISAT2, or Salmon for gene isoform counting and extensive quality control of the sequenced data. The major bottlenecks in RNA-Seq data analysis include manual installations of software, deployment platforms, or computational capacity and cost.
Looking at the vast number of tools available for a single analysis and different versions and their compatibility makes the setup tricky. This can also be time-consuming as proper configuration and version compatibility assessment take several months to complete.
Nextflow: Solution to Bottleneck
The most efficient way to tackle these hurdles is by making use of Nextflow based pipelines that support cloud computing where virtual systems can be provisioned at a fraction of the cost, and the setup is seemingly smoother that can be done by a single individual, as well as support for container systems (Docker and Singularity).
Nextflow is a reactive workflow framework and a programming DSL (Domain Specific Language) that eases the writing of data-intensive computational pipelines.
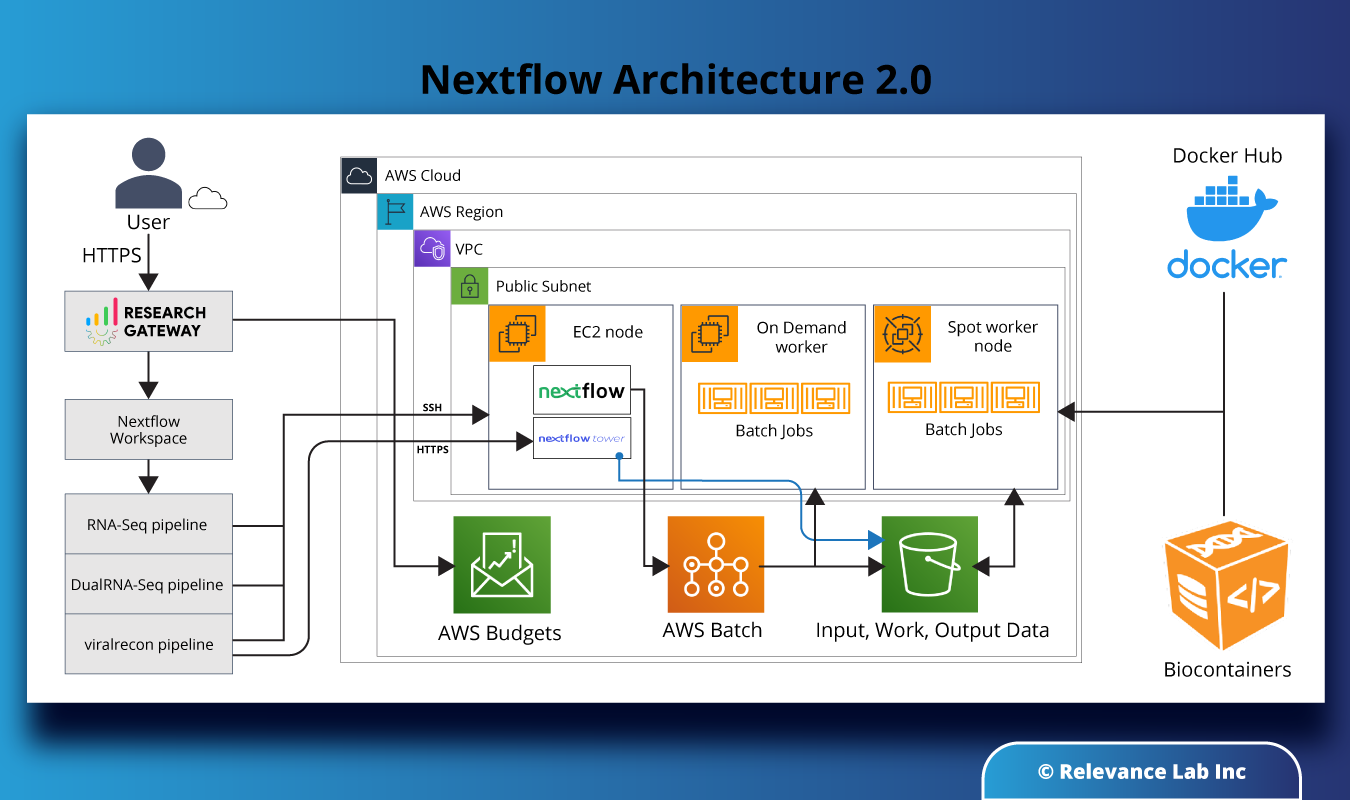
As seen in the diagram below, the infrastructure to use Nextflow in the AWS cloud consists of a head node (EC2 instance with Nextflow and Nextflow Tower open source software installed) and wired to an AWS Batch backend to handle the tasks created by Nextflow. AWS Batch creates worker nodes at run-time, which can be either on-demand instances or spot instances (for cost-efficiency). Data is stored in an S3 bucket to which the worker nodes in AWS Batch connect and pull the input data. Interim data and results are also stored in S3 buckets, as is the output. The pipeline to be run (e.g. RNA-Seq, DualRNA-Seq, ViralRecon, etc.) is pulled by the worker nodes as a container image from a public repo like DockerHub or BioContainers.
RLCatalyst Research Gateway takes care of provisioning the infrastructure (EC2 node, AWS Batch compute environment, and Job Queues) in the AWS cloud with all the necessary controls for networking, access, data security, and cost and budget monitoring. Nextflow takes care of creating the job definitions and submitting the tasks to Batch at run-time.
The researcher initiates the creation of the workspace from within the RLCatalyst Research Gateway portal. There is a wide selection of parameters as input including, which pipeline to run, tuning parameters to control the sizing and cost-efficiency of the worker nodes, the location of input and output data, etc. Once the infrastructure is provisioned and ready, the researcher can connect to the head node via SSH and launch Nextflow jobs. The researcher can also connect to the Nextflow Tower UI interface to monitor the progress of jobs.
The pre-written Nextflow pipelines can be pulled from an nf-core GitHub repository and can be set up within minutes allowing the entire analysis to run using a single line command, and the results of each step are displayed on the command line/shell. Configuration of the resources on the cloud is seamless as well, since Nextflow based pipelines provide support for batch computing, enabling the analysis to scale as it progresses. Thus, the researchers can focus on running the pipeline and analysis of output data instead of investing time in setup and configurations.
As seen from the pipeline output (MultiQC) report of the Nextflow-based RNA-Seq pipeline below, we can identify the sequence quality by looking at FastQC scores, identify duplication scenarios based on the contour plots as well as pinpoint the genotypic biotypes along with fragment length distribution for each sample.
RLCatalyst Research Gateway enables the setting up and provisioning of AWS cloud resources with few simple clicks for such analysis, and the output of each run is saved in a S3 bucket enabling easy data sharing. These provisioned resources are pre-configured setups with a proper design template and security architecture and added to these features. RLCatalyst Research Gateway enables cost tracking for the currently running projects, which can be paused/ stopped or deleted as per convenience.
Steps for Running Nextflow-Based Pipelines in AWS Cloud for Genomic Research
Prerequisites for a researcher before starting data analysis.
A valid AWS account and access to the RLCatalyst Research Gateway portal
A publicly accessible S3 bucket with large Research Data sets accessible
Once done, below are the steps to execute this use case.
Login to the RLCatalyst Research Gateway Portal and select the project linked to your AWS account
Launch the Nextflow-Advanced product
Login to the head node using SSH (Nextflow software will already be installed on this node)
In the pipeline folder, modify the nextflow.config file to set the data location according to your needs (Github repo, S3 bucket, etc.). This can also be passed via the command line
Run the Nextflow job on the head node. This should automatically cause Nextflow to submit jobs to the AWS Batch backend
Output data will be copied to the Output bucket specified
Once done, terminate the EC2 instance and check for the cost spent on the use case
All costs related to the Nextflow project and researcher consumption are tracked automatically
Key Points
Bioinformatics involves developing methodology and analysis tools to analyze large volumes of biological data
Vast number of tools available for a single analysis and their compatibility make the analysis setup tricky
RLCatalyst Research Gateway enables the setting up and provisioning of Nextflow based pipelines and AWS cloud resources with few simple clicks
Summary
Researchers need powerful tools for collaboration and access to commonly used NGS pipelines with large data sets. Cloud computing makes it much easier with access to workflows, data, computation, and storage. However, there is a learning curve for researchers to use Cloud-specific knowhow and how to use resources optimally for large-scale computations like RNA-Seq analysis pipelines that can also be quite costly. Relevance Lab working closely with AWS partnership has provided RLCatalyst Research Gateway portal to use commonly used pre-built Nextflow pipeline templates and integration with open source repositories like nf-core and biocontainers. RLCatalyst Research Gateway enables execution of such Nextflow-based scalable pipelines on the cloud with few clicks and configurations with cost tracking and resource execution control features. By using AWS Batch the solution is very scalable and optimized for on-demand consumption.
We use cookies on our website to provide you with a more relevant experience. To learn more about how we use cookies and how you can manage your cookie settings, please refer to our Privacy.
We do not collect and sell your personal information.
This website uses cookies to improve your experience while you navigate through the website. Out of these cookies, the cookies that are categorized as necessary are stored on your browser as they are essential for the working of basic functionalities of the website. We also use third-party cookies that help us analyze and understand how you use this website. These cookies will be stored in your browser only with your consent. You can opt-out of non- necessary cookies. But opting out of some of these cookies may have an effect on your browsing experience.
Necessary cookies are absolutely essential for the website to function properly. This category only includes cookies that ensures basic functionalities and security features of the website. These cookies do not store any personal information.